The concepts of pharmaceutical equivalence and therapeutic equivalence
Fedaisf editorial staff
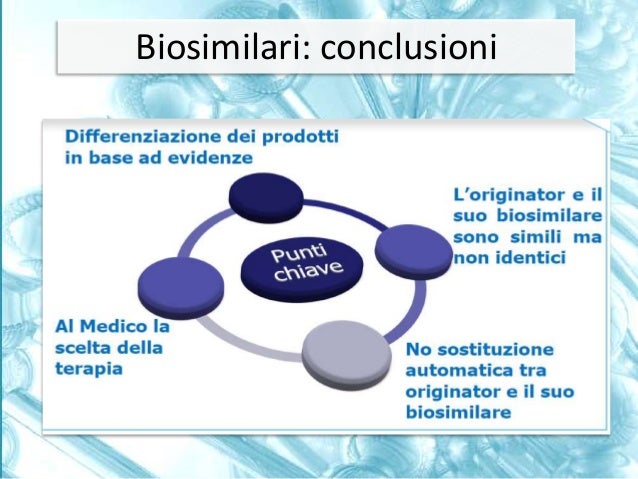
From pharmaceutical equivalence one cannot go directly to therapeutic equivalence. The only way to obtain a direct demonstration of therapeutic equivalence is to conduct phase 3 studies of a comparative nature. The generic drug can be approved without the need for direct demonstration of therapeutic equivalence
An interview with Corrado Blandizzi Full Professor of Pharmacology, Department of Clinical and Experimental Medicine, University of Pisa
Pharmaceutical equivalence and therapeutic equivalence: can you give us a definition and explain the differences?
These definitions apply to processes in which comparisons are made between different pharmaceutical products that contain the same active ingredient. We speak of pharmaceutical equivalence when two products, with the same type of formulation, contain the exact same active ingredient, but differ in terms of excipients according to certain parameters predefined by regulatory activities. From pharmaceutical equivalence it is not possible to pass directly to therapeutic equivalence, because the latter depends on the type of drug on which we intend to apply these criteria. If it concerns the approval of a generic drug, the Regulatory Authority asks to take a further step, that is to pass from pharmaceutical equivalence to bioequivalence. This is obtained with a pharmacokinetic study on healthy volunteers, which makes it possible to demonstrate that the times and concentrations with which the active ingredients of both products first reach the blood and then the target organs do not differ significantly. Once the criteria of pharmaceutical equivalence and bioequivalence are met, the generic drug can be approved without the need for direct demonstration of therapeutic equivalence. The only way to obtain a direct demonstration of therapeutic equivalence is to conduct phase 3 studies of a comparative nature. This stage is necessary for biosimilar drugs, for which a preliminary study is envisaged aimed at demonstrating their similarity in a chemical, physical, biological, and I would even say pharmacological context, with the original product. Having established that this similarity or, better to say, biosimilarity exists, it can be requested that a judgment of therapeutic equivalence be expressed by the Regulatory Authority no longer on the basis of bioequivalence studies, but on the basis of clinical studies before phase 1 and then above all phase 3, which must demonstrate the non-inferiority of the biosimilar product compared to the original product both from the point of view of therapeutic efficacy and from the point of view of safety of use.
The market entry of biosimilars of complex molecules is imminent. What are, in your opinion, the conditions that could guarantee the maximum protection of the patient's health?
Since central authorization is granted not only for therapeutic indications for which there is clinical evidence, but also for those for which the indication is exclusively extrapolated in the absence of clinical evidence, access to the market, and therefore to clinical practice, of new biosimilar drugs of complex molecules such as monoclonal antibodies can give rise to some concern not only in healthcare professionals but also in patients. This could be remedied by determining a more controlled process of accessing the drug to the market, allowing it to be initially used only in accredited centers which should record all its effects in terms of efficacy and safety, and then re-evaluate - after a certain number of years (from two to five) - the actual behavior of the drug in particular with respect to the extrapolated indications, to assess whether there is room for liberalizing the use of the new biosimilar drug even in a context in which the indication had been extrapolated .
ML
Biosimilar drugs. The role of the prescriber
Si è rimarcata l’evidente situazione di scollamento tra gli enti regolatori e i clinici, che si devono confrontare ogni giorno con la responsabilità penale individuale di prescrivere un farmaco in una condizione nella quale nulla è scritto in letteratura.
A colloquio con Giovanni Lapadula Professore Ordinario di Reumatologia, Università di Bari e Direttore dell’Unità Operativa di Reumatologia dell’Azienda Universitaria-Ospedaliera di Bari
Let's talk about extrapolation: what is the current implementation from a regulatory point of view and what are the implications for the prescriber?
Il concetto di estrapolazione è stato molto ben chiarito sul piano tecnico dagli enti regolatori. In base a questo concetto si afferma che lì dove si documenti un’efficacia simile di un farmaco rispetto ad alcune indicazioni, si può allargarne l’indicazione anche ad altre patologie, con la possibilità – prevista dai Position Paper degli enti regolatori come FDA, EMA e AIFA – che si faccia una valutazione indicazione per indicazione, eventualmente attivando un approfondimento delle possibilità di fare l’estrapolazione dei dati. Questi buoni propositi degli enti regolatori (e soprattutto di EMA, che è quella che ci riguarda direttamente) sono stati puntualmente smentiti nel momento in cui è stato approvato il primo farmaco biosimilare, per il quale – senza alcuna valutazione aggiuntiva – si è fatta un’estrapolazione a tutte le indicazioni che aveva il farmaco originatore. Questo procedimento ha creato sconcerto tra i medici prescrittori, perché queste decisioni sono state prese senza consultare chi ha un’esperienza diretta della terapia sul singolo individuo. Si è così rimarcata l’evidente situazione di scollamento tra gli enti regolatori e i clinici, che si devono confrontare ogni giorno con la responsabilità penale individuale di prescrivere un farmaco in una condizione nella quale nulla è scritto in letteratura. Questo è il grosso problema, di cui non si è tenuto alcun conto, e che esprime una difficoltà nei rapporti tra le diverse anime dei professionisti che si occupano della cura e della salute dei pazienti.
Cos’è l’INN e quali le implicazioni rispetto al tema della tracciabilità?
L’INN è un numero che identifica una molecola. Ogni molecola, diversa da un’altra, ha un numero differente. A questo proposito emerge ancora una volta da parte degli enti regolatori, questa volta a livello mondiale, un segnale di scarsa considerazione delle necessità di chi opera sul territorio e deve prescrivere la terapia e sorvegliare sui suoi effetti. Gli enti regolatori hanno infatti deciso di utilizzare un unico numero identificativo sia per il farmaco biologico originatore sia per i biosimilari, i quali però – per definizione – non sono uguali ma ‘biosimilari’. Hanno quindi degli elementi di diversità, anche se minimi, per i quali non sono indicabili come identici e avrebbero meritato un INN individuale. Questo non si è voluto fare, complicando moltissimo la tracciabilità dei farmaci. Nel momento in cui compare un evento avverso, il medico dovrebbe infatti essere messo in grado di individuare la molecola che lo ha determinato, per evitare di attribuire gli effetti collaterali di un farmaco ad un altro. Si è deciso invece di ricorrere nelle segnalazioni all’indicazione del nome commerciale del farmaco, determinando anche la necessità di ristrutturare i database dei registri, che in passato prevedevano l’indicazione delle molecole e non del nome commerciale. Il problema è particolarmente complesso per la variabilità intrinseca dei farmaci biotecnologici, per i quali occorrerebbe indicare il numero di lotto di appartenenza. Su questo aspetto però non si è fatta chiarezza, perché di fatto esistono segnalazioni degli eventi avversi eseguite con il nome commerciale, ma non c’è nessuna segnalazione del numero di lotti coinvolti, perché i clinici non sono stati avvertiti che questo è un elemento importante dal quale dipende la salute dei nostri pazienti. Bisogna infatti considerare che il momento registrativo dei farmaci biosimilari si è limitato a sperimentazioni di sei mesi su numeri di pazienti fra le io e le 5o volte più bassi rispetto alle sperimentazioni sui farmaci originatori, in maniera assolutamente legale, ma che comunque non ha convinto del tutto chi poi deve prescrivere il farmaco e vorrebbe poterlo fare in piena sicurezza per il paziente.