
Un nostro lettore ci segnala il convegno organizzato dalla Federazione nazionale della Stampa italiana e dall’Istituto superiore di sanità, in collaborazione con Web Health Information Network (Whin), venerdì 15 giugno scorso, nella sede dell’Iss come corso di formazione per giornalisti dedicato al tema dei ‘Farmaci generici’.
Nel corso si è sviluppato e sviscerato le varie problematiche dei farmaci generici/equivalenti fra cui ‘Le bufale sui farmaci equivalenti’, a cura di Annalisa Manduca. Alla fine si è tenuta la tavola rotonda: “Comunicare la salute. Le esperienze dei giornalisti del settore”, a cui hanno  partecipato Maria Emilia Bonaccorso, Carla Massi, Annalisa Manduca, Elisa Manacorda, Mauro Boldrini e Carla Bruschelli.
partecipato Maria Emilia Bonaccorso, Carla Massi, Annalisa Manduca, Elisa Manacorda, Mauro Boldrini e Carla Bruschelli.
La Dr.ssa Bruschelli ha lasciato un’intervista sui generici. In questa intervista dichiarava che questi farmaci vengono sottoposti a controllo rigoroso da parte di AIFA, parla di un 20% in più o in meno degli eccipienti, e del risparmio da loro prodotto da reinvestire nella ricerca.
Ora ci dispiace immensamente dover correggere la Dr.ssa Bruschelli per la quale abbiamo stima e rispetto.
Nel nostro paese, i farmaci generici sono definiti in base alla Legge di conversione n.425 del 8.8.1996, testo coordinato del Decreto legge 20.6.1996 n.323 – GU n.191 del 16.8.1996, come Medicinali a base di uno o più principi attivi, prodotti industrialmente, non protetti da brevetto o certificato protettivo complementare (CPC), identificati dalla denominazione comune internazionale del principio attivo (API), seguita dal nome del titolare della AIC. Inoltre, sono distinti in generici branded [o specialità analoghe] e generici unbranded [principio attivo + nome produttore]. Tali farmaci sono stati poi ridefiniti come Medicinali equivalenti (Legge 149 del 26 luglio 2005), dato che il termine “generico” finiva per essere percepito dall’utilizzatore come simile piuttosto che uguale al farmaco di riferimento.
 Detto questo a proposito della legge, veniamo alle sue dichiarazioni.
Detto questo a proposito della legge, veniamo alle sue dichiarazioni.
Vi sono alcuni importanti aspetti inerenti la qualità del principio attivo generico e dei suoi eccipienti rispetto alla molecola originale (es. presenza di impurità, sterilità della soluzione ricostituita, ecc.) che dovrebbero essere presi in considerazione nella valutazione tecnica del generico prima della sua introduzione in commercio (AIC). Infatti l’AIC si basa su l’autocertificazione del produttore o del paese da cui proviene. Purtroppo questa autocertificazione non garantisce alcunché. È recente il caso del valsartan in cui erano presenti impurità potenzialmente cancerogene dal 2012 e nessuno s’è accorto di nulla, prima c’era stato il caso della messa al bando da parte di EMA di 300 generici e prima ancora di 700 farmaci generici prodotti in India e poi il ritiro degli 80 in Germania, e così via. Con questo non vogliamo certo dire che i generici sono pericolosi. la maggior parte di essi sono sicuri. e gli eventuali inquinanti riscontrati non sono comunque dannosi.
La normativa prevede che ci sia la stessa quantità di principio attivo, ma è la biodisponibilità che può variare da +20 a -20% rispetto all’originatore, non gli eccipienti, ne può risultare quindi che due generici posti agli estremi del range possono variare fra loro del 40%, forse un po’ troppo. Manca la definizione dei singoli farmaci e/o delle classi di farmaci a ristretto indice terapeutico (quelli per cui una minima differenza può comportare sensibili variazioni di efficacia e sicurezza). Non esistono studi per dimostrare l’equivalenza terapeutica, di efficacia e sicurezza (si suppone soltanto che ci sia, a parità di molecola), negli USA per esempio c’è L’Orange Book che lo fa.
Il farmaco generico può essere approvato senza necessità di una dimostrazione diretta di equivalenza terapeutica. L’unico modo per ottenere una dimostrazione diretta di equivalenza terapeutica sarebbe condurre studi di fase 3 di natura comparativa.
 Gli eccipienti possono variare ed incidono in modo fondamentale sulla biodisponibilità, soprattutto in alcuni preparati, come per esempio le soluzioni, le gocce oculari, ecc., in cui possono alterare viscosità, osmolarità e pH nei colliri oculari con un evidente impatto su tollerabilità e penetrazione oculare.
Gli eccipienti possono variare ed incidono in modo fondamentale sulla biodisponibilità, soprattutto in alcuni preparati, come per esempio le soluzioni, le gocce oculari, ecc., in cui possono alterare viscosità, osmolarità e pH nei colliri oculari con un evidente impatto su tollerabilità e penetrazione oculare.
Non esiste quindi certezza scientifica sul fatto che una volta comprovata la bioequivalenza tra farmaco brand e composto generico sussegua una paragonabile tollerabilità ed efficacia ovvero equivalenza terapeutica.
Venendo al risparmio è da considerare che è la scadenza di brevetto a far crollare il prezzo cioè la fine di un monopolio sul principio attivo dell’originatore. Questo dovrebbe innescare una concorrenza dei prezzi fra l’originatore e i suoi generici. Ciò avviene solo in parte perché l’originatore deve per legge avere un prezzo superiore al generico. Inoltre il prezzo più basso del generico diventa il prezzo di riferimento che il SSN rimborsa, la differenza di prezzo degli altri farmaci uguali sarà a carico del paziente. A queste condizioni il risparmio dello Stato è inesistente. Cioè non c’è alcun risparmio per la spesa sanitaria territoriale essendoci un prezzo di riferimento.Facendo riferimento alla concorrenza, il “libero mercato” è impedito anche dallo sconto, per legge, con cui i farmaci generici sono acquistati dai farmacisti. Sconto superiore rispetto agli originali, cioè il farmacista percentualmente ci guadagna di più con il generico. Infine se col brevetto c’era un regime di monopolio del farmaco di marca, con la scadenza dello stesso si passa ad un “mercato” dominato da un oligopolio di sole 5 aziende di generici.
La banalizzazione del prezzo non prevede una visione a lungo termine, ma individuano in un unico capro espiatorio tutti i mali di un settore. Oggi è la volta del farmaco. Ci sembra che si accavallano le posizioni interessate di Assogenerici con le posizioni del SSR e SSN come efficace opera di distrazione.
Dati presenti nella letteratura medico-scientifica internazionale, alcune esperienze condotte in Italia e le segnalazioni di reazioni avverse ai farmaci presenti nel database dell’AIFA (compresi i casi di inefficacia terapeutica) concorrono a sostenere l’ipotesi che nei mercati farmaceutici di vari Paesi (compresa l’Italia) accanto a farmaci equivalenti di buona qualità siano presenti anche farmaci equivalenti di scarsa qualità. Senza norme rigorose il “cattivo” generico scredita tutta la categoria dei generici.
Ci sarebbe molto altro da dire, ma non vogliamo annoiare i nostri lettori.
Gent.le Dr. Bruschelli, siamo oltremodo imbarazzati a farle notare queste inesattezze da lei dette e, dato che partecipa anche a programmi televisi, la invitiamo cortesemente a documentarsi un po’ meglio.
Redazionale – 6 agosto 2018
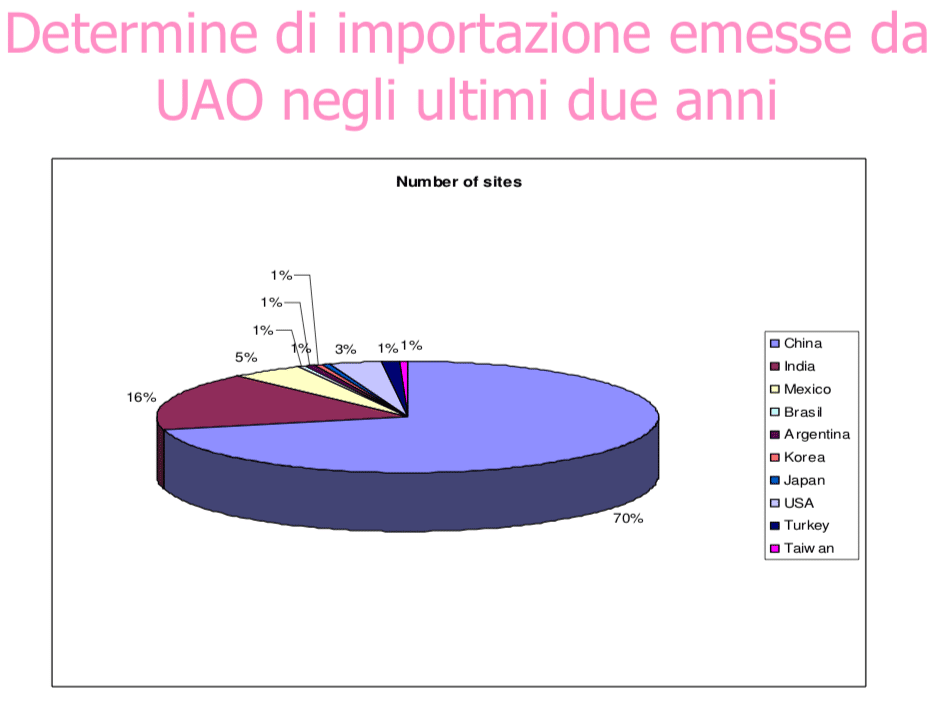
Nota: UAO: Ufficio Autorizzazioni Officine
API (Active Pharmaceutical Ingredient) principi attivi
Notizie correlate: L’Aifa ritira lotti di farmaci generici contenenti un potenziale cancerogeno: diciamo tutta la verità!
Farmaci generici: tra opportunità e falsi miti





