
From pharmaceutical equivalence one cannot go directly to therapeutic equivalence. The only way to obtain a direct demonstration of therapeutic equivalence is to conduct phase 3 studies of a comparative nature. The generic drug can be approved without the need for direct demonstration of therapeutic equivalence
CARE Care costs and economic resources
An interview with Corrado Blandizzi Full Professor of Pharmacology, Department of Clinical and Experimental Medicine, University of Pisa
Pharmaceutical equivalence and therapeutic equivalence: can you give us a definition and explain the differences?
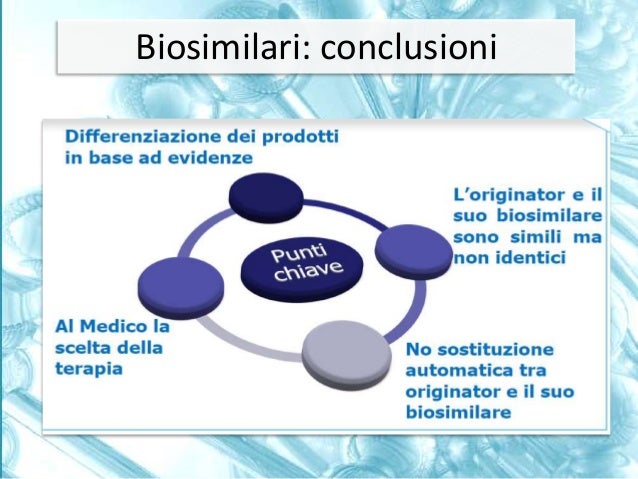
 These definitions apply to processes in which comparisons are made between different pharmaceutical products that contain the same active ingredient. We speak of pharmaceutical equivalence when two products, with the same type of formulation, contain the exact same active ingredient, but differ in terms of excipients according to certain parameters predefined by regulatory activities. From pharmaceutical equivalence it is not possible to pass directly to therapeutic equivalence, because the latter depends on the type of drug on which we intend to apply these criteria. If it concerns the approval of a generic drug, the Regulatory Authority asks to take a further step, that is to pass from pharmaceutical equivalence to bioequivalence. This is obtained with a pharmacokinetic study on healthy volunteers, which makes it possible to demonstrate that the times and concentrations with which the active ingredients of both products first reach the blood and then the target organs do not differ significantly. Once the criteria of pharmaceutical equivalence and bioequivalence are met, the generic drug can be approved without the need for direct demonstration of therapeutic equivalence. The only way to obtain a direct demonstration of therapeutic equivalence is to conduct phase 3 studies of a comparative nature. This stage is necessary for biosimilar drugs, for which a preliminary study is envisaged aimed at demonstrating their similarity in a chemical, physical, biological, and I would even say pharmacological context, with the original product. Having established that this similarity or, better to say, biosimilarity exists, it can be requested that a judgment of therapeutic equivalence be expressed by the Regulatory Authority no longer on the basis of bioequivalence studies, but on the basis of clinical studies before phase 1 and then above all phase 3, which must demonstrate the non-inferiority of the biosimilar product compared to the original product both from the point of view of therapeutic efficacy and from the point of view of safety of use.
These definitions apply to processes in which comparisons are made between different pharmaceutical products that contain the same active ingredient. We speak of pharmaceutical equivalence when two products, with the same type of formulation, contain the exact same active ingredient, but differ in terms of excipients according to certain parameters predefined by regulatory activities. From pharmaceutical equivalence it is not possible to pass directly to therapeutic equivalence, because the latter depends on the type of drug on which we intend to apply these criteria. If it concerns the approval of a generic drug, the Regulatory Authority asks to take a further step, that is to pass from pharmaceutical equivalence to bioequivalence. This is obtained with a pharmacokinetic study on healthy volunteers, which makes it possible to demonstrate that the times and concentrations with which the active ingredients of both products first reach the blood and then the target organs do not differ significantly. Once the criteria of pharmaceutical equivalence and bioequivalence are met, the generic drug can be approved without the need for direct demonstration of therapeutic equivalence. The only way to obtain a direct demonstration of therapeutic equivalence is to conduct phase 3 studies of a comparative nature. This stage is necessary for biosimilar drugs, for which a preliminary study is envisaged aimed at demonstrating their similarity in a chemical, physical, biological, and I would even say pharmacological context, with the original product. Having established that this similarity or, better to say, biosimilarity exists, it can be requested that a judgment of therapeutic equivalence be expressed by the Regulatory Authority no longer on the basis of bioequivalence studies, but on the basis of clinical studies before phase 1 and then above all phase 3, which must demonstrate the non-inferiority of the biosimilar product compared to the original product both from the point of view of therapeutic efficacy and from the point of view of safety of use.
The market entry of biosimilars of complex molecules is imminent. What are, in your opinion, the conditions that could guarantee the maximum protection of the patient's health?
Since central authorization is granted not only for therapeutic indications for which there is clinical evidence, but also for those for which the indication is exclusively extrapolated in the absence of clinical evidence, access to the market, and therefore to clinical practice, of new biosimilar drugs of complex molecules such as monoclonal antibodies can give rise to some concern not only in healthcare professionals but also in patients. This could be remedied by determining a more controlled process of accessing the drug to the market, allowing it to be initially used only in accredited centers which should record all its effects in terms of efficacy and safety, and then re-evaluate - after a certain number of years (from two to five) - the actual behavior of the drug in particular with respect to the extrapolated indications, to assess whether there is room for liberalizing the use of the new biosimilar drug even in a context in which the indication had been extrapolated .
ML
Biosimilar drugs. The role of the prescriber
The clear disconnection between regulatory bodies and clinicians was highlighted, who are faced every day with the individual penal responsibility of prescribing a drug in a condition in which nothing is written in the literature.
Ca|Re Regioni (Care) – Published by Redazione – 26/01/2015
An interview with Giovanni Lapadula Full Professor of Rheumatology, University of Bari and Director of the Rheumatology Operative Unit of the University Hospital of Bari
Let's talk about extrapolation: what is the current implementation from a regulatory point of view and what are the implications for the prescriber?
 The concept of extrapolation has been very well clarified on a technical level by the regulatory bodies. On the basis of this concept it is stated that where a similar efficacy of a drug is documented with respect to some indications, the indication can also be extended to other pathologies, with the possibility – provided for by the Position Papers of regulatory bodies such as the FDA, EMA and AIFA – that an evaluation be made indication by indication, possibly activating an in-depth study of the possibilities of extrapolating data. These good intentions of the regulatory bodies (and above all of EMA, which is the one that directly concerns us) were duly denied when the first biosimilar drug was approved, for which - without any additional evaluation - an extrapolation to all indications that the originator drug had. This procedure has created confusion among prescribing physicians, because these decisions were made without consulting those who have direct experience of the therapy on the individual. Thus the evident disconnection between regulatory bodies and clinicians was highlighted, who are faced every day with the individual penal responsibility of prescribing a drug in a condition in which nothing is written in the literature. This is the big problem, of which no account has been taken, and which expresses a difficulty in the relationships between the different souls of the professionals who deal with the care and health of patients.
The concept of extrapolation has been very well clarified on a technical level by the regulatory bodies. On the basis of this concept it is stated that where a similar efficacy of a drug is documented with respect to some indications, the indication can also be extended to other pathologies, with the possibility – provided for by the Position Papers of regulatory bodies such as the FDA, EMA and AIFA – that an evaluation be made indication by indication, possibly activating an in-depth study of the possibilities of extrapolating data. These good intentions of the regulatory bodies (and above all of EMA, which is the one that directly concerns us) were duly denied when the first biosimilar drug was approved, for which - without any additional evaluation - an extrapolation to all indications that the originator drug had. This procedure has created confusion among prescribing physicians, because these decisions were made without consulting those who have direct experience of the therapy on the individual. Thus the evident disconnection between regulatory bodies and clinicians was highlighted, who are faced every day with the individual penal responsibility of prescribing a drug in a condition in which nothing is written in the literature. This is the big problem, of which no account has been taken, and which expresses a difficulty in the relationships between the different souls of the professionals who deal with the care and health of patients.
What is the INN and what are the implications with respect to traceability?
 The INN is a number that identifies a molecule. Each molecule, different from another, has a different number. In this regard, once again the regulatory bodies, this time at a global level, show a sign of scant consideration of the needs of those who work in the area and who have to prescribe the therapy and monitor its effects. In fact, the regulatory bodies have decided to use a single identification number both for the originator biological drug and for the biosimilars, which however – by definition – are not the same but 'biosimilars'. They therefore have elements of diversity, even if minimal, for which they cannot be indicated as identical and would have deserved an individual INN. This was not done, greatly complicating the traceability of drugs. When an adverse event occurs, the doctor should in fact be able to identify the molecule that caused it, to avoid attributing the side effects of one drug to another. Instead, it was decided to use the indication of the commercial name of the drug in the reports, also determining the need to restructure the register databases, which in the past provided for the indication of the molecules and not the commercial name. The problem is particularly complex due to the intrinsic variability of biotechnological drugs, for which it would be necessary to indicate the lot number to which they belong. However, there was no clarity on this aspect, because in fact there are reports of adverse events performed under the trade name, but there is no report of the number of lots involved, because clinicians have not been warned that this is an important element on which the health of our patients depends. In fact, it must be considered that the time of registration of biosimilar drugs was limited to six-month trials on numbers of patients between 10 and 50 times lower than trials on the originator drugs, in an absolutely legal manner, but which in any case did not convince the all those who then have to prescribe the drug and would like to be able to do so in complete safety for the patient.
The INN is a number that identifies a molecule. Each molecule, different from another, has a different number. In this regard, once again the regulatory bodies, this time at a global level, show a sign of scant consideration of the needs of those who work in the area and who have to prescribe the therapy and monitor its effects. In fact, the regulatory bodies have decided to use a single identification number both for the originator biological drug and for the biosimilars, which however – by definition – are not the same but 'biosimilars'. They therefore have elements of diversity, even if minimal, for which they cannot be indicated as identical and would have deserved an individual INN. This was not done, greatly complicating the traceability of drugs. When an adverse event occurs, the doctor should in fact be able to identify the molecule that caused it, to avoid attributing the side effects of one drug to another. Instead, it was decided to use the indication of the commercial name of the drug in the reports, also determining the need to restructure the register databases, which in the past provided for the indication of the molecules and not the commercial name. The problem is particularly complex due to the intrinsic variability of biotechnological drugs, for which it would be necessary to indicate the lot number to which they belong. However, there was no clarity on this aspect, because in fact there are reports of adverse events performed under the trade name, but there is no report of the number of lots involved, because clinicians have not been warned that this is an important element on which the health of our patients depends. In fact, it must be considered that the time of registration of biosimilar drugs was limited to six-month trials on numbers of patients between 10 and 50 times lower than trials on the originator drugs, in an absolutely legal manner, but which in any case did not convince the all those who then have to prescribe the drug and would like to be able to do so in complete safety for the patient.




