For a drug to receive authorization from the regulatory agency to be placed on the market, it is necessary for the manufacturing company to demonstrate its efficacy, quality and safety. However, the assessment of the level of security is often  very complex and requires additional procedures that go beyond the clinical trial phase of the drug. "The safety [of a medicine] is not absolute and can only be judged in relation to its efficacy," reads a document on the subject produced by the World Health Organization.
very complex and requires additional procedures that go beyond the clinical trial phase of the drug. "The safety [of a medicine] is not absolute and can only be judged in relation to its efficacy," reads a document on the subject produced by the World Health Organization.
“There is a possibility that rare but serious adverse reactions may go undetected during drug development” [1]. For this reason, part of the safety evaluation process of a drug must necessarily take place after it has been approved for sale, when the drug is already on the market. This phase, defined as "post-marketing surveillance", does not only concern innovative medicines in the approval phase but also generics and medicines already approved in the past and on the market.
How is the safety of a drug evaluated?
In order for its use as a medicine to be authorized, a molecule must undergo a long series of experimental studies, first in the laboratory and on animals and finally on humans. On average, the total duration of the process of
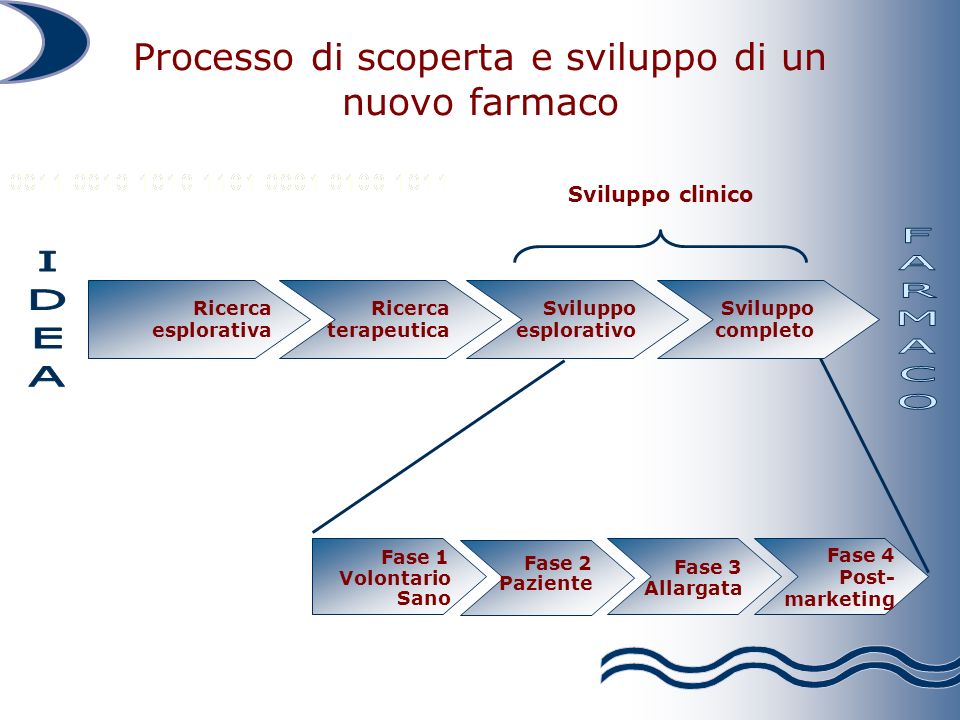
marketing authorization for a drug varies between seven and ten years.
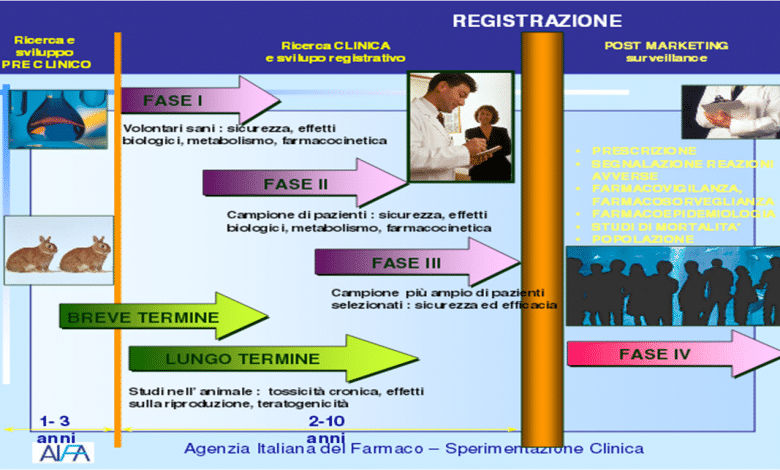
The first phase of experimentation is defined as pre-clinical as it does not involve the administration of the molecule to human subjects: its behavior (route of administration, absorption, elimination) and level of toxicity are evaluated on laboratory samples and animal models.
Only if the drug proves to have a potential therapeutic effect and an acceptable level of toxicity does it pass to the clinical trial phase, consisting of three different phases. In the first, the molecule is tested, in various dosages, on a limited number of young and healthy patients, in order to evaluate any side effects that did not emerge in the pre-clinical phase. In the second phase, however, the drug is tested on volunteer subjects (usually from 100 to 300 subjects, but in some cases even more) affected by the disease for which the drug is designed, and is therefore functional to demonstrate the non-toxicity and the activity of the active ingredient. Finally, in the third and final phase, the efficacy and safety of the molecule are evaluated compared to a placebo, another drug or no treatment, on samples generally made up of thousands of subjects [2].
What is Post-Marketing Surveillance?
As mentioned above, however, some adverse reactions may not occur during the drug's experimental phases - as they are particularly rare - or may simply occur later or in subgroups of patients not included in the studies. This problem emerged dramatically in the late 1950s and early 1960s  when it was realized that the administration of the anti-nausea drug thalidamide to pregnant women caused severe deformities in the unborn children [1].
when it was realized that the administration of the anti-nausea drug thalidamide to pregnant women caused severe deformities in the unborn children [1].
Even if the problem of adverse reactions to drugs was already known previously, from that moment the institutions understood the need to implement a series of coordinated interventions to evaluate the emergence of these situations and act promptly to resolve them [3].
This "fourth phase", therefore, has the objective of evaluating those adverse reactions that can be identified only after the mass use of a drug and is precisely called "post-marketing surveillance". The results of these analyzes are then used to update the package insert of the medicine or, in the most serious cases, to proceed with the cancellation of the authorization by the regulatory agency.
Doctor… but how does it work?
In Italy, the holder of the Marketing Authorization (MA) of the drug, usually the manufacturing company, has - according to the legislative decree 219/2006, which regulates these mechanisms – the obligation to “register and notify with the  utmost urgency, and in any case within fifteen days of receiving news of it, any suspected serious adverse reaction from medicinal products, which occurred in Italy and reported to him by healthcare personnel, to the healthcare facility to which the reporter belongs and, where it is not possible to identify this facility, to the 'Italian Medicines Agency (AIFA)" [4].
utmost urgency, and in any case within fifteen days of receiving news of it, any suspected serious adverse reaction from medicinal products, which occurred in Italy and reported to him by healthcare personnel, to the healthcare facility to which the reporter belongs and, where it is not possible to identify this facility, to the 'Italian Medicines Agency (AIFA)" [4].
Spontaneous reports from doctors and pharmacists represent the largest part of communications on potential adverse reactions, but they are not the only ones. In many cases, for example, real epidemiological studies are organized which allow, in addition to identifying any unexpected adverse reactions, also to evaluate their frequency [1]: in this type of research, for example, it is identified - through the prescription data – a population of patients undergoing a specific treatment and contacting them (e.g. by submitting a questionnaire) to ascertain any adverse reactions [5].
Author “The Scientific Thought” – July 3, 2018
Related news: Can a drug not be “effective” even if it worked for me?




