
“Non possiamo accettare i commenti di chi allude a un mercato italiano su cui si aggirano prodotti non sottoposti ad adeguati controlli o dotati di standard di qualità inferiori a quelli previsti dalla legge, tanto più che questi stessi farmaci subiscono tutti i controlli di legge per ottenere l’autorizzazione all’immissione in commercio”
HealthDesk – 28 settembre 2019
Le aziende produttrici di farmaci equivalenti (i cosiddetti “generici”) guardano «con grande attenzione e rispetto all’attività di controllo sui medicinali svolta dalle Agenzie regolatorie internazionali per garantire la massima sicurezza dei medicinali utilizzati dai cittadini. La richiesta dell’Agenzia europea Ema di sottoporre a test precauzionali tutti i farmaci in commercio in Europa risponde esattamente a questa esigenza e dimostra che il sistema dei controlli è efficace».

Enrique Häusermann, presidente di Assogenerici, commenta così il dibattito scatenato in questi giorni dalla richiesta dell’Ema
«Ema è alleata delle aziende farmaceutiche nel garantire i massimi standard di qualità e sicurezza della produzione – assicura Häusermann – e le fake news fanno più danni alla salute di qualsiasi potenziale impurezza se portano alla sospensione delle terapie».
Le nitrosamine sono sostanze naturalmente presenti in natura e sono ritenute potenzialmente cancerogene se assunte in quantità elevatissime per un lungo periodo di tempo, cosa che non accade con l’assunzione delle terapie farmacologiche, anche per il trattamento continuativo nei pazienti cronici.
La scelta dell’Ema di sospendere l’utilizzo della ranitidina e di chiedere alle aziende di testare tutti i medicinali in commercio «risponde al principio di massima precauzione – osserva il presidente Assogenerici – e testimonia la qualità e il rigore del meccanismo dei controlli applicati a tutto il settore farmaceutico e  condivisi a livello comunitario. Farmacovigilanza e attività di ispezione, verifica e analisi nel comparto farmaceutico sono in continua evoluzione: vengono ispezionati i siti produttivi, vengono seguiti test prima, durante e dopo la produzione».
condivisi a livello comunitario. Farmacovigilanza e attività di ispezione, verifica e analisi nel comparto farmaceutico sono in continua evoluzione: vengono ispezionati i siti produttivi, vengono seguiti test prima, durante e dopo la produzione».
Häusermann assicura che «metteremo in campo ogni sforzo attenendoci alle disposizioni dettate dall’Agenzia regolatoria» e si augura che «l’argomento non venga sfruttato ad arte per gettare ombre sul settore o per scatenare falsi allarmi. Non possiamo accettare i commenti di chi allude a un mercato italiano su cui si aggirano prodotti non sottoposti ad adeguati controlli o dotati di standard di qualità inferiori a quelli previsti dalla legge, tanto più che questi stessi farmaci subiscono tutti i controlli di legge per ottenere l’autorizzazione all’immissione in commercio. Allusioni o velate insinuazioni sulla correttezza delle procedure utilizzate dai produttori – prosegue Häusermann – gettano un’ombra offensiva anche sull’attività svolta dalle Agenzie regolatorie, a partire dall’Agenzia italiana del farmaco Aifa, che è invece perfettamente allineata all’Ema nell’attività di vigilanza controllo su efficacia, qualità e sicurezza dei prodotti in commercio che – vale la pena di ricordarlo – sono gli stessi in tutta Europa».
HealthDesk – 28 settembre 2019
N.d.R.: Chissà, forse il problema è proprio la legge che accetta autocertificazioni o certificazioni dei paesi produttori. Esiste poi un problema di bioequivalenza terapeutica. Cosa che in Italia si dà per scontato fra generici farmacologicamente equivalenti, ma che scontato non è. In Europa ed in Italia non c’è per questo problema l’equivalente dell’Orange Book americano. Perché?
Riportiamo un estratto della prefazione dell’Orange Boook:
In generale, ai prodotti farmaceutici che l’Agenzia (FDA) considera multisorgente è stato assegnato un codice di equivalenza terapeutica. Il sistema di codifica per le valutazioni dell’equivalenza terapeutica è  progettato per consentire agli utenti di determinare rapidamente se l’Agenzia ha valutato un particolare prodotto approvato come terapeuticamente equivalente ad altri prodotti farmaceuticamente equivalenti (prima lettera) e fornire ulteriori informazioni sulla base delle valutazioni della FDA (seconda lettera). Con alcune eccezioni (ad es. Valutazioni di equivalenza terapeutica per alcune domande di commercializzazione semplificate secondo la sezione 505 (b) della norma FDA)
progettato per consentire agli utenti di determinare rapidamente se l’Agenzia ha valutato un particolare prodotto approvato come terapeuticamente equivalente ad altri prodotti farmaceuticamente equivalenti (prima lettera) e fornire ulteriori informazioni sulla base delle valutazioni della FDA (seconda lettera). Con alcune eccezioni (ad es. Valutazioni di equivalenza terapeutica per alcune domande di commercializzazione semplificate secondo la sezione 505 (b) della norma FDA)
( )), la data di valutazione dell equivalenza terapeutica è la stessa della data di approvazione. Le due categorie di base in cui sono stati inseriti farmaci multisorgente sono indicate dalla prima lettera del relativo codice di equivalenza terapeutica come segue:
A: Prodotti farmaceutici che la FDA considera terapeuticamente equivalenti ad altri prodotti farmaceuticamente equivalenti, ovvero prodotti farmaceutici per i quali:
( ) non esistono problemi di bioequivalenza noti o sospetti. Questi sono designati come AA, AN, AO, AP o AT, a seconda della forma di dosaggio; o
( ) i problemi di bioequivalenza effettivi o potenziali sono stati risolti con adeguate prove in vivo e / o in vitro a supporto della bioequivalenza. Questi sono designati AB.
B: Prodotti farmaceutici che la FDA in questo momento considera non terapeuticamente equivalenti ad altri prodotti farmaceuticamente equivalenti, vale a dire prodotti farmaceutici per i quali i problemi di bioequivalenza reali o potenziali non sono stati risolti da prove adeguate di bioequivalenza. Spesso il problema è con forme di dosaggio specifiche piuttosto che con i principi attivi. Questi sono indicati come BC, BD, BE, BN, BP, BR, BS, BT, BX o B. I singoli prodotti farmaceutici sono stati valutati come terapeuticamente equivalenti al prodotto di riferimento in conformità con le definizioni e le politiche FDA.
I prodotti farmaceutici designati con un codice “B” rientrano in una delle tre politiche principali:
( ) i prodotti farmaceutici contengono ingredienti attivi o sono fabbricati in forme di dosaggio che sono state identificate dall Agenzia come aventi problemi di bioequivalenza documentati o un potenziale significativo per tali problemi e per i quali non sono stati presentati alla FDA studi adeguati che dimostrino la bioequivalenza; o
( ) gli standard di qualità sono inadeguati o la FDA ha una base insufficiente per determinare l’equivalenza terapeutica; o
 ( ) i prodotti farmaceutici sono sottoposti a revisione normativa. (Estratto da Orange Book Preface)
( ) i prodotti farmaceutici sono sottoposti a revisione normativa. (Estratto da Orange Book Preface)
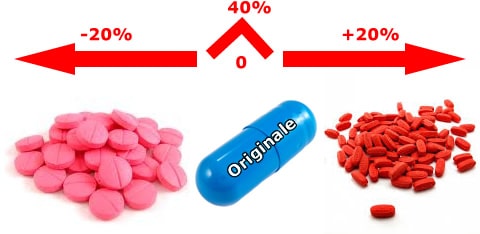
In Italia una delle questioni dei generici riguarda l’equivalenza di farmaci generici tra loro. Poiché i test di bioequivalenza non sono condotti anche tra farmaci equivalenti dello stesso prodotto di “marca”, ma solo tra quest’ultimo e ogni singolo equivalente, e poiché la bioequivalenza non gode della proprietà transitiva, né il medico né il farmacista hanno informazioni sufficienti per confrontare tra loro i prodotti equivalenti (bio-creep). In un mercato sempre più aperto alla produzione dei generici, è evidente che la mancanza di confrontabilità tra generici è un ostacolo alla loro sostituibilità e all’impegno dei medici e dei farmacisti di scegliere il farmaco tra tutti quelli, equivalenti, in commercio. Inoltre, la non confrontabilità comporta anche la difficoltà per il medico di scegliere, tra gli equivalenti, il prodotto che più si avvicina, per intervallo di confidenza, a quello originale.




