
Siringhe, alcuni tipi di sciroppo, mascherine e laser chirurgici, apparecchiature di laboratorio, ma anche bisturi, valvole cardiache e protesi d’anca: fanno tutti parte della categoria dei dispostivi medici per i quali è in corso una rivoluzione senza precedenti: dal 26 maggio 2021, infatti, è in vigore il nuovo “Regolamento Europeo Dispositivi Medici” che modifica le norme che disciplinano questi prodotti. L’obiettivo è garantire un elevato livello di sicurezza, tracciabilità e protezione della salute favorendo l’innovazione. Il comparto dei dispositivi medici complessivamente genera un mercato che vale 16,7 miliardi di euro tra export e mercato interno e conta 4.323 aziende di produzione e distribuzione, che occupano 94.153 dipendenti. Di queste 2.523 sono aziende di produzione. Il mondo dei dispositivi medici conta circa 1,5 milioni di tecnologie per la salute e il benessere delle persone attualmente in commercio.
Le aziende hanno tempo fino al 26 maggio 2024 per adeguarsi alle nuove regole e ‘ri-certificare’ i vari device. Ma fino ad ora soltanto una minoranza ha regolarizzato i propri prodotti. Un ritardo che rischia di rendere problematici gli approvvigionamenti. Per sensibilizzare aziende, istituzioni e cittadini sul tema, l’Accademia per la Salute e la Ricerca clinica lancia la campagna di sensibilizzazione ‘SOS Dispositivi medici: in meta entro il 2024’ realizzata con il contributo non condizionato di CROLife, organizzazione con un team specialistico che fornisce servizi di supporto alle aziende che operano nel campo medico per adempiere alcuni dei compiti insiti nelle fasi di uno studio clinico migliorando l’efficienza in tutti i suoi processi.
In base alle vecchie direttive (direttiva 90/385/CEE, direttiva 93/42/CEE) per ottenere la marcatura CE, il produttore doveva dimostrare che il dispositivo fornisse le prestazioni per cui era stato progettato e che i rischi prevedibili e la frequenza degli eventuali eventi avversi fossero ridotti al minimo accettabile. Se i dati a supporto di quanto richiesto non erano sufficienti, veniva raccomandato lo svolgimento di ulteriori indagini cliniche.
Il nuovo Regolamento prevede espressamente che per ottenere la conformità ai requisiti essenziali di sicurezza e prestazione, le aziende produttrici debbano effettuare una valutazione clinica dei dati disponibili, ed eventualmente, se i dati non sono sufficienti, predisporre ulteriori indagini cliniche. «Il bisturi, la valvola cardiaca e la protesi d’anca sono dispositivi medici, così come tutti gli strumenti chirurgici utilizzati normalmente in sala operatoria dai chirurghi, cardiochirurghi e ortopedici»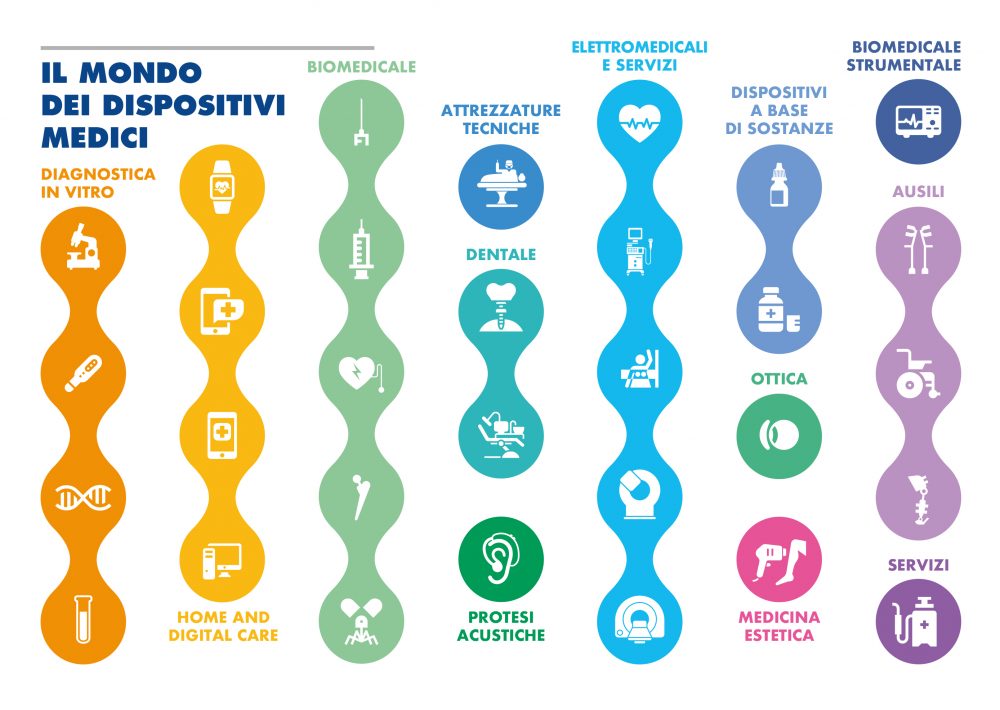 , spiega Marcella Marletta, già direttore generale del Ministero della Salute e ora direttore generale dell’Accademia per la Salute e Ricerca clinica oltre che docente presso l’Università Guglielmo Marconi di Roma e presso l’Università Campus BioMedico. «Tutti i dispositivi medici sono regolamentati dal nuovo Regolamento 2017/745 in base al quale entro il 26 maggio 2024, fine del periodo transitorio, molte imprese di dispositivi medici dovranno certificare i propri prodotti per i quali sono scaduti i certificati CE di immissione in commercio e molte aziende dovranno ri-certificare prodotti che sono soggetti ad una nuova classificazione».
, spiega Marcella Marletta, già direttore generale del Ministero della Salute e ora direttore generale dell’Accademia per la Salute e Ricerca clinica oltre che docente presso l’Università Guglielmo Marconi di Roma e presso l’Università Campus BioMedico. «Tutti i dispositivi medici sono regolamentati dal nuovo Regolamento 2017/745 in base al quale entro il 26 maggio 2024, fine del periodo transitorio, molte imprese di dispositivi medici dovranno certificare i propri prodotti per i quali sono scaduti i certificati CE di immissione in commercio e molte aziende dovranno ri-certificare prodotti che sono soggetti ad una nuova classificazione».
Le valutazioni e le indagini cliniche dei Dispositivi Medici ai sensi del nuovo Regolamento Europeo 745 (MDR) rappresentano alcune delle prossime sfide più grandi per i fabbricanti europei per garantire la conformità di prodotti efficaci e sicuri. «I fabbricanti dovrebbero applicare le nuove norme, insieme ai documenti di orientamento dell’UE, per valutare la rispondenza di ogni singolo prodotto ai nuovi paradigmi», spiega Fernanda Gellona, direttore generale di Confindustria dispositivi medici.  «In caso di veri e propri gap nelle evidenze occorrerà sfruttare al meglio il tempo rimanente per intraprendere un appropriato follow-up clinico post-commercializzazione (PMCF) e ridurre tali lacune in vista del cambio di passo richiesto dalla certificazione ai sensi dell’MDR.
«In caso di veri e propri gap nelle evidenze occorrerà sfruttare al meglio il tempo rimanente per intraprendere un appropriato follow-up clinico post-commercializzazione (PMCF) e ridurre tali lacune in vista del cambio di passo richiesto dalla certificazione ai sensi dell’MDR.![]() Laddove ciò non risultasse sufficiente, occorrerà generare nuove evidenze tramite indagini e valutazioni cliniche a supporto della destinazione d’uso del dispositivo».
Laddove ciò non risultasse sufficiente, occorrerà generare nuove evidenze tramite indagini e valutazioni cliniche a supporto della destinazione d’uso del dispositivo».
La certificazione dei device è in mano agli Organismi notificati. Ad oggi in Europa ce ne sono 27 e fino ad ora hanno certificato solo 500 dispositivi delle migliaia presenti sul mercato. «Ad oggi le imprese sono in difficoltà a farsi ri-certificare prodotti in scadenza o a certificarne di nuovi anche a causa della diminuita capacità di certificazione da parte degli Organismi notificati», sottolinea la dott.ssa Marletta che aggiunge: «Si tratta di un grandissimo problema perché gli Organismi notificati deputati alle nuove certificazioni sono assolutamente insufficienti».
Dunque, il nuovo Regolamento genera un aumento dei costi per le imprese, la produzione di ulteriori evidenze cliniche, l’inasprimento dei controlli di vigilanza e sorveglianza post-vendita. Con quali conseguenze per le aziende produttrici? «E’ chiaro che – risponde la dottoressa Gellona – le nuove regole renderanno il mercato più selettivo sul piano della sicurezza e della qualità, aspetto senz’altro positivo; al tempo stesso, però, la complessità del sistema farà aumentare notevolmente i tempi per le certificazioni e dunque per rendere disponibili le tecnologie innovative. Inoltre, i costi connessi alle nuove regole renderanno molto più difficile l’accesso al mercato soprattutto per le PMI. Tutta questa situazione potrebbe comportare una generale perdita di competitività europea rispetto ad altri mercati globali, uno spostamento degli investimenti per nuovi prodotti verso alcuni mercati esteri come quelli americano e cinese ed infine il ritiro di determinati prodotti o causare fusioni e acquisizioni di convenienza».
Secondo una survey condotta da Medtech Europe, associazione europea delle aziende di dispositivi medici, l’80% dei fabbricanti sta incontrando difficoltà nell’avvio o nel completamento del percorso di conformità e pensa di non certificare il 15-20% dei dispositivi. Alcuni medi e piccoli produttori potrebbero uscire dal mercato e dal sistema. «Il bisturi “xy”, ben conosciuto dal chirurgo che si è formato professionalmente e lo ha utilizzato per tanti anni per operare con la migliore tecnica i suoi pazienti, potrebbe sparire dal commercio e non essere più disponibile», avverte Marletta. La stessa cosa potrebbe accadere con la protesi d’anca e con tanti altri dispositivi medici indispensabili per la nostra salute.
Questa situazione di ‘stallo’ preoccupa gli addetti ai lavori, ma potrebbe avere anche delle conseguenze dirette sui pazienti perché restano soltanto due anni per mettersi in regola con le nuove disposizioni del Regolamento Europeo sui dispositivi medici. «In base ai dati dell’Associazione europea degli Organismi notificati – dichiara Fernanda Gellona – si stima che i certificati in scadenza nella prima metà del 2024 siano 7272. Questo significa che ci sarà un picco nel carico di lavoro degli Organismi notificati che sarà difficile da gestire.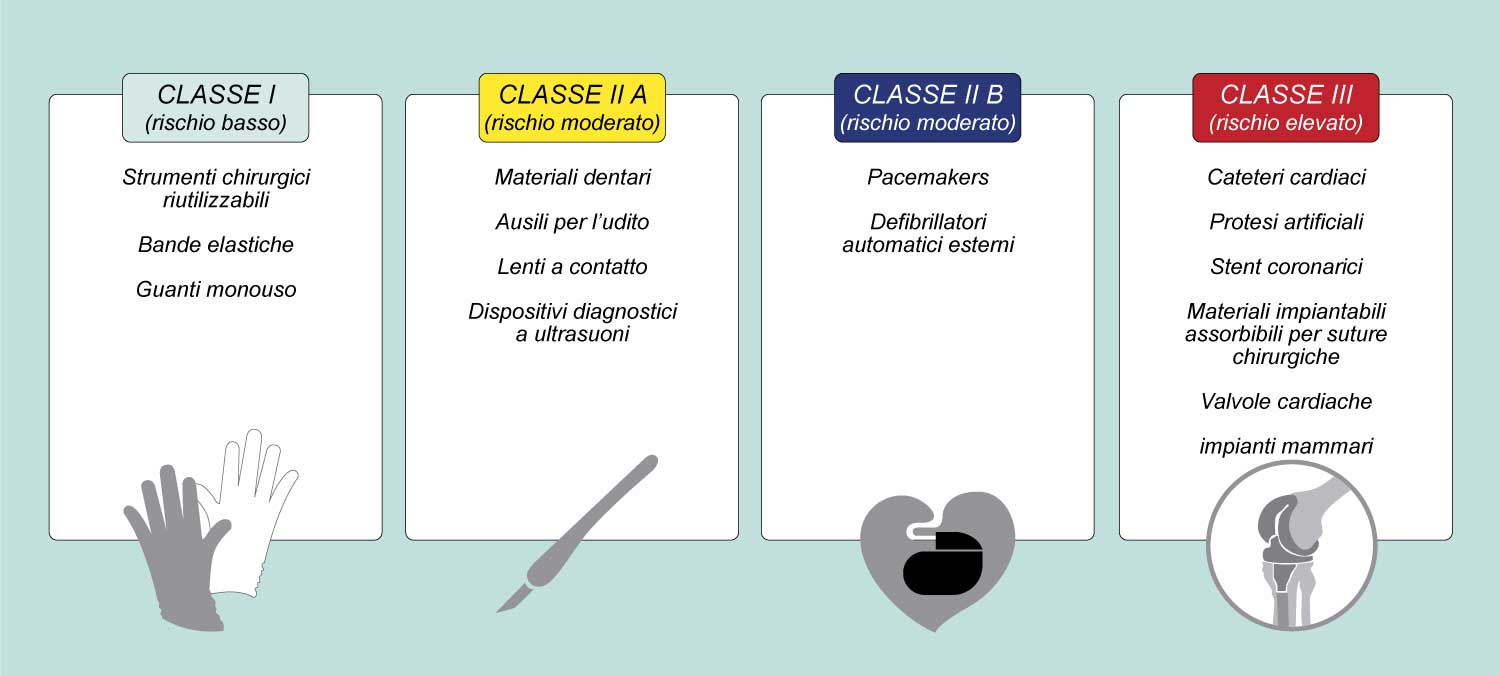 Tenendo conto della durata del processo di certificazione, potrebbero non riuscire ad essere certificati ai sensi del nuovo regolamento per la fine del periodo di grazia. Il rischio che alcuni dispositivi potrebbero non essere disponibili esiste per due motivi: la mancanza di un numero sufficiente di organismi notificati e la scelta da parte delle imprese di certificare solo alcuni dispositivi. È fondamentale in questo momento che tutte le parti interessate collaborino affinché si possa mettere le aziende nelle giuste condizioni per poter completare il nuovo processo certificativo ai sensi dell’MDR e, quindi, sia possibile continuare a mettere a disposizione dei pazienti cure efficaci e sicure», raccomanda la dottoressa Gellona.
Tenendo conto della durata del processo di certificazione, potrebbero non riuscire ad essere certificati ai sensi del nuovo regolamento per la fine del periodo di grazia. Il rischio che alcuni dispositivi potrebbero non essere disponibili esiste per due motivi: la mancanza di un numero sufficiente di organismi notificati e la scelta da parte delle imprese di certificare solo alcuni dispositivi. È fondamentale in questo momento che tutte le parti interessate collaborino affinché si possa mettere le aziende nelle giuste condizioni per poter completare il nuovo processo certificativo ai sensi dell’MDR e, quindi, sia possibile continuare a mettere a disposizione dei pazienti cure efficaci e sicure», raccomanda la dottoressa Gellona.
Ma qual è attualmente la situazione in Italia? «Da una ricerca nella banca dati del Ministero della salute, da me condotta nel 2020, nel mio ruolo di Direttore Generale, al fine di rilevare la situazione in Italia – risponde Marletta – è stato avviato un censimento presso gli 11 Organismi Notificati italiani, volto a conoscere l’attività ed in particolare il numero dei certificati in scadenza che per vari motivi non sarebbero stati rinnovati e i Dispositivi Medici che potevano venire a mancare, nonché le richieste di certificazioni di nuovi prodotti rifiutate per indisponibilità e già risultava che 320 certificati erano in scadenza». In base ai dati di un’altra ricerca, ancora in corso, ci sarebbero più di 15.000 certificati rilasciati in base alle vecchie direttive da trasformare in certificati in regola con quanto disposto dal nuovo Regolamento MDR da qui al 2024.
Molte aziende, grandi e piccole, oggi sono ancora spaesate dal nuovo Regolamento e non sapendo bene quali passi compiere si affidano ad una Contract Research Organization.  Quali requisiti e competenze deve avere una CRO? «In primo luogo – risponde la dottoressa Marletta – è fondamentale che lo staff in grado di effettuare una valutazione critica dei pro e contro di qualsiasi studio clinico. Poi serve una conoscenza approfondita della normativa di riferimento per poter presentare tutta la documentazione alle Agenzie Regolatorie in modo celere e con un alto standard di qualità. Inoltre, le CRO più qualificate interagiscono continuamente con molti ospedali e investigatori, quindi possono raccomandare siti in grado di reclutare il tipo di pazienti necessari in uno specifico studio».
Quali requisiti e competenze deve avere una CRO? «In primo luogo – risponde la dottoressa Marletta – è fondamentale che lo staff in grado di effettuare una valutazione critica dei pro e contro di qualsiasi studio clinico. Poi serve una conoscenza approfondita della normativa di riferimento per poter presentare tutta la documentazione alle Agenzie Regolatorie in modo celere e con un alto standard di qualità. Inoltre, le CRO più qualificate interagiscono continuamente con molti ospedali e investigatori, quindi possono raccomandare siti in grado di reclutare il tipo di pazienti necessari in uno specifico studio».
Per mantenere alta l’attenzione su questo tema e stimolare stakeholders, aziende, istituzioni e anche l’opinione pubblica, l’Accademia per la Salute e Ricerca clinica lancia la campagna di sensibilizzazione ‘SOS Dispositivi medici: in meta entro il 2024’ con una serie di eventi online dedicati all’approfondimento di questi temi a cui sarà possibile partecipare gratuitamente. Il primo appuntamento è il 26 maggio – anniversario dell’entrata in vigore del nuovo Regolamento europeo – alle ore 18 sulla piattaforma Microsoft Teams di CroLife con Fernanda Gellona e Marcella Marletta. Per iscriversi a questo evento gratuito, basta andare su https://www.crolife.eu/register-to-special-event/ e compilare il modulo di registrazione.
Fonte: Panorama della Sanità – 19 maggio 2022
—————————————————————————————————-
Nota:
I regolamenti UE sui dispositivi sono stati emanati entrambi nel 2017
– Regolamento 2017/ 745 / UE – MDR medical device regulation – entrato in vigore il 25 maggio 2017 ed al quale è stata riconosciuto un periodo di “transizione” di 3 anni, poi con il Reg. UE 2020/ 561 prorogato di un altro anno. Applicabile a decorrere dal 25 maggio 2021. * Sintesi
-Regolamento 2017 / 746 / UE – IVDR in vitro diagnostic regulation – transizione di 5 anni. Applicabile quindi dal 26 maggio 2022.
Il 21 dicembre 2021 Katia Accorsi, Presidente di Assodiagnostici di Confindustria dispositivi medici, che valuta positivamente la recente approvazione da parte del Consiglio e del Parlamento Europeo dell’ importate modifica all’IVDR, che entrerà comunque in vigore il 26 maggio (2022 ndr), ma permetterà alle aziende operanti nella produzione e distribuzione di test diagnostici in vitro di adeguare i propri prodotti tra il 2024 e il 2028 a seconda del tipo di classificazione del device, così commenta: “L’approvazione dell’emendamento relativo al nuovo regolamento sui dispositivi medico-diagnostici in vitro (IVDR), grazie al quale verranno estesi i periodi transitori, rappresenta un’ottima prova di dialogo e cooperazione tra le istituzioni a livello europeo e nazionale”.
Il Regolamento (UE) 2022/112 del Parlamento europeo e del Consiglio del 25 gennaio 2022 ha modificato il Regolamento (UE) 2017/746 in relazione alle disposizioni transitorie per determinati dispositivi medico-diagnostici in vitro e l’applicazione differita delle condizioni concernenti i dispositivi fabbricati e utilizzati all’interno della stessa istituzione sanitaria (“dispositivi in house”).
Per approfondire:
Linee Guida europea sui prodotti borderline – Ministero della Salute – 19 maggio 2022
Istituzione della rete nazionale per la vigilanza – Ministero della Salute – 2 maggio 2022
Nuovi regolamenti europei sui dispositivi medici – PandsLegal 2019
I regolamenti europei sui dispositivi medici – Ministero della Salute agg. 3 feb 2022




